

I løbet af de sidste årtier af min karriere har jeg brugt utallige timer på at arbejde for at beskytte amerikanere ved at forske i lægemidlers sikkerhed. Min uddannelse og karriere har ført mig gennem omkring et halvt dusin universiteter, Big Pharma, og hos FDA under tre præsidentielle administrationer. Lægemiddelsikkerhed overvejer, hvorfor en person kan tage et farmaceutisk produkt og have nul bivirkninger, mens en anden person kan tage det samme produkt, men har bivirkninger op til og inklusive permanent invaliditet eller død. Som standard overvejer undersøgelser af lægemiddelsikkerhed også ikke-kliniske aspekter af fremstilling og lægemiddelkvalitet.
Fordi lægemiddelkvalitet er en væsentlig faktor i vurderingen af lægemiddelsikkerhed, førte min vandring for at beskytte amerikanerne til min konceptualisering og grundlæggelse af verdens første "analytisk apotek” missioneret med videnskabeligt at verificere farmaceutiske produkter fra steder som Indien og Kina, før de udleveres til patienter. Desværre førte jagten på stort over etik og beskyttelse af patienter til, at virksomhedens økonomiske ledelse forpligtede sig omfattende FDA-overtrædelser og bliver beskyldt af dommere for at lave falske videnskabelige påstande (hvilket ved et uheld skete efter min exit).
Uden ekstern bekræftelse af lægemiddelkvalitet er amerikanerne fuldstændig afhængige af FDA og producenter for at vurdere og bekræfte produktets renhed. Lægemiddelsikkerhed har vist sig at være et bemærkelsesværdigt problem, når det kommer til Covid mRNA-injektioner. Desværre, hvis nogen ønskede at udføre deres egen analyse af mRNA-injektioner, de ikke har en passende detaljeret ingrediensliste at sammenligne den med, eller endda adgang til den etablerede reguleringsmetode for, hvordan man korrekt tester den for renhed.
Det er fordi producenterne , FDA overvejer alle ingredienser i disse mRNA-injektioner, inklusive sekvensen af mRNA plus lipid nanopartikel (LNP) egenskaber, inklusive halveringstid, LNP strukturer, overflademodifikation(er), antal/type(r) af LNP'er pr. dosis og vedhæftningspunkter på mRNA-strengen, for at være uspecificeret eller "forretningshemmelighed".
Derudover overvejer FDA desuden metoder om, hvordan man tester mRNA-injektioner for renhed også en forretningshemmelighed.
Bipartisan støtte og hundredvis af milliarder af skatteydere, men INGEN gennemsigtighed?
Covid-mRNA-hemmelighed eksisterer, selvom både Trump- og Biden-administrationerne havde foreslået fuld gennemsigtighed med mRNA-injektioner til det punkt, hvor Covid-mRNA-intellektuelle ejendomsrettigheder ophæves. På trods af det tillader/holder både FDA og producenterne patenter, herunder grundlæggende data om disse skud, som en forretningshemmelighed. Det gør de på trods af, at alle Covid-vaccineproducenter har modtaget hundreder af millioner af skatteydere efter Forbes/Statista publikationer.
At studere narkotikasikkerhedsepidemiologi er svært nok. Uden verificerbar produktrenhed/konsistens er en komplet sikkerhedsvurdering umulig.
Fuld gennemsigtighed af alle ingredienser og kvalitetskontrolforanstaltninger er vigtige, ikke kun fordi de var stærkt skatteyderfinansieret med hundredvis af millioner af dollars, men fordi der er opstået en række spørgsmål om sikkerheden og effektiviteten af Covid mRNA-injektioner.
Ud over at være usædvanligt komplekse, blev deres godkendelse fremskyndet af regulatorer efter mindre end et år. De fleste lægemidler og vacciner tager typisk rundt ti år for fuldt ud at teste for sikkerhed/effektivitet og gennemgå og godkende. Ud over at ingredienserne er helt nye, meget komplekse og de første af sin slags, der skal administreres i massiv skala, udvikler bl.a. langsigtede kliniske sikkerheds-/toksicitetsevalueringer og epidemiologiske undersøgelser blev fremskyndet og sandsynligvis ikke fuldt ud belyst før frigivelsen.
FDA-ingrediensbekræftelse, gennemsigtighed og "sandhed" har præcedenser, der går tilbage til 1800-tallet:
Den analytiske verifikation og gennemsigtigheden af ingredienser eller "sandheden i mærkning", hvor indholdet af flasken er påkrævet for at matche de angivne ingredienser går forud for oprettelsen af FDA, tilbage til 1862. Dagens FDA blev faktisk født ud af, hvad der startede som en enkelt "Department of Chemistry"-medarbejder ansat i det amerikanske landbrugsministerium.
forfalskning, (ændrede eller giftige ingredienser) forkert mærke (indeholder en falsk etiket eller er på anden måde vildledende eller indeholder ukorrekte medicinske påstande), eller fejlmærkning (indeholder ingrediens(er) er ikke anført på et produktmærke) har alle haft lange, grimme historier i Amerika. Det blev troet, at den uhyggelige karakter havde toppet i begyndelsen til midten af det 19. århundrede - eller i det mindste da det blev identificerbart - da først i 1862 var tekniske processer blevet udviklet til at analysere og opdage svindel med ingredienser. Inden da ville såkaldte "rejsende medicinmænd", der kalder sig "læger" (uvægerligt med tvivlsomme eller ikke-eksisterende akkreditiver), slynge flasker med "helbredende" produkter, hvis ingrediensetiketter kun ville vise tåget eller uskadeligt indhold som f.eks. “vitaminer""urteekstrakter,"Eller"slangeolie” – eller har ofte slet ingen ingrediensliste.
Dengang mange fromme, puritanske New Englanders, som af religiøse årsager ville aldrig røre ved alkohol, ville købe disse opløsninger fra disse smygende hucksters og ubevidst blive narret til at forbruge opløsninger, som ikke kun indeholdt alkohol, men narkotika som opium og/eller kokain. Under påskud af at forbedre et uhyggeligt bredt overflødighedshorn af lidelser, udviklede patienter i stedet straffende afhængighed og/eller på anden måde fik deres helbred negativt påvirket af disse tidlige "narkohandlere".
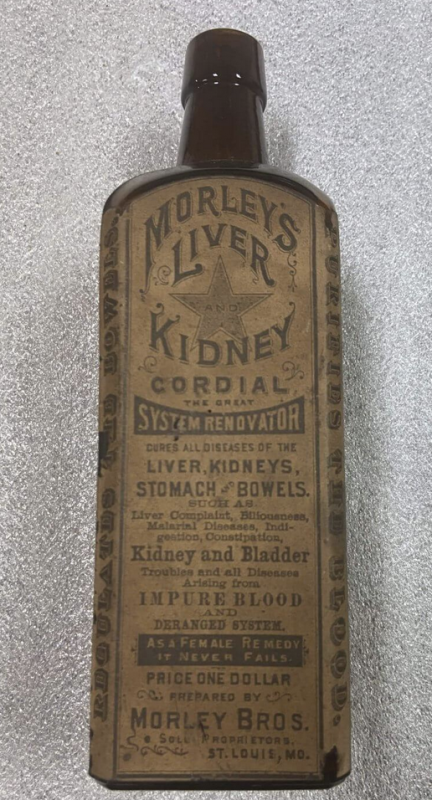
Efterhånden som problemet voksede, begyndte den føderale regering at lægge mærke til det. Til sidst, den Lov om ren mad og stoffer blev vedtaget i 1906 og førte til oprettelsen af Food and Drug Administration (FDA).
[FDA havde en formativ pligt til at sikre, at lægemidler bærer sandfærdige mærkningserklæringer og opfylder visse standarder for renhed og styrke.
Husk den næsten 120-årige sandfærdigt mærkningskrav og "renhed" del af Pure Food and Drug Act af 1906, mens du læser videre om mRNA-verifikationstest og ingrediensgennemsigtighed.]
Hvilken "sandfærdig" og "ren" ingrediensverifikationstest finder sted for FDA-regulerede produkter?
Tilbage i 2021 valgte FDA at begynde at overvåge USAs farmaceutiske kvalitet via en fjernindsamling of indsendt indsendelse af prøver for stoffer som erstatning for inspektioner af levende anlæg på grund af Covid-pandemien. Var det lovligt? Kunne det nogensinde blive betragtet som videnskabeligt passende? I dag, på trods af at pandemien er afsluttet, er den eneste officielle farmaceutiske frigivelsestest, der i øjeblikket udføres på enhver Covid mRNA lægemiddel kommer til syne til stadig udføres af FDA via en producentleveret, "indsendt” prøve i henhold til a skærmbillede af den aktuelle FDA-hjemmeside. Det er klart, at en "indsendt" prøveudtagningsmetode er meget anderledes og potentielt mindre pålidelig end direkte indsamling af prøver via en direkte, personlig indsamlingsmetode. På trods af det hævder FDA, at det har "den højeste standard over hele kloden for prøveudtagning og testning".
Desuden foreslår FDA yderligere at fremme sin "indsendte" fjerntestpolitik med en nyligt foreslået vejledningsdokument.
Selvom det kun eksisterer som et "udkast" til FDA-dokument, viser officielle FDA-websteder det indsendelsen af prøver ser ud til allerede at have været implementeret siden mindst januar 2021. FDA ser ud til at hævde resultaterne af disse indsendte tests som deres uafhængige verifikation.
Desuden er det nederst på første side af FDA-udkastet dokumentet foreslår udvidelse af "fjerntest". Det lister i øjeblikket hver FDA produktregulerende afdeling hos FDA, hvilket antyder, at det er et agenturdækkende politikforslag.
Den komplette liste inkluderer:
- Kontoret for Regulatoriske Anliggender
- Kontoret for fødevarepolitik og -reaktion
- Kontor for kombinationsprodukter
- Center for Biologisk Evaluering og Forskning
- Center for Lægemiddelvurdering og -forskning
- Center for Apparater og Radiologisk Sundhed
- Center for Fødevaresikkerhed og Anvendt Ernæring
- Center for Tobaksvarer
- Center for Veterinærmedicin
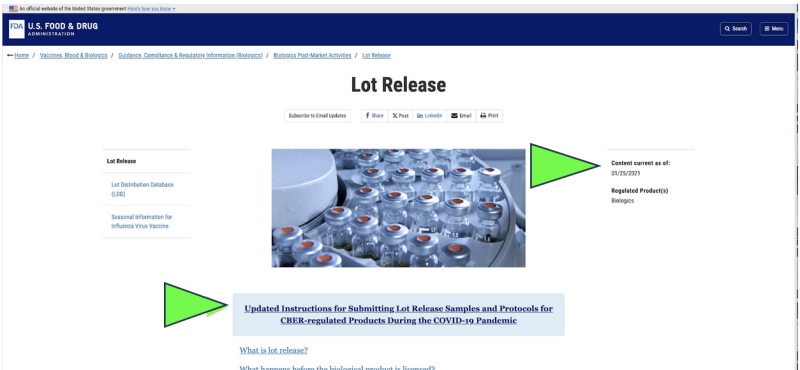
Er "indsendt" kvalitetskontrolprøver af FDA passende? Hvad hvis statens sundhedsafdelings restaurantinspektioner afspejlede FDA-politik?
Denne "mail-in" prøveudtagningsmetode er på samme måde absurd, for eksempel for en stats sundhedsafdeling, der overvåger restauranter ved at bede dem om periodisk at "maile" forskellige varer fra deres menu til en testfacilitet, så sundhedsafdelingerne kan teste for potentiel mad -født forurening og/eller beder restauranter om at love selv at teste menupunkter. Hvad hvis den restaurant var i Kina? Hvad hvis den restaurant var i Indien? Eller ethvert andet land, der er kendt for at have en afgrundsdyb historie med svindel og kvalitetskontrol problemer?
Denne metode ville være uacceptabel for både restauranter og medicinalvirksomheder, af årsager, som omfatter det indlysende: fabrikanter kunne indsende de prøver, de foretrækker – ikke nødvendigvis repræsentative batchprøver. Det er åbenbart ikke det samme som at FDA-inspektører erhverver prøver under uanmeldte inspektioner af hele anlægget.
Under restaurantanalogien ville selvfølgelig alle restauranter indsende prøver af karakteren "A". hvilket ikke nødvendigvis ville være repræsentativt for, hvad forbrugerne modtager.

Kvalitetskontrol: Hvad er farmaceutisk "frigivelsestest", og hvorfor er det vigtigt?
I dag fører FDA tilsyn med kvaliteten og indholdet af $2.7 billioner værdi af produktet årligt, men ser ud til at undertrykke kritiske ingrediensverifikationsvurderinger og resultater. FDA formodes at beskytte amerikanere ved at udføre omfattende analytisk test som en kontrolsum for at sikre ingrediensnøjagtighed. Resultaterne heraf bør være gennemsigtige for skatteyderne, der finansierer FDA's $6.6 milliarder budget. Denne videnskabelige verifikation omtales som farmaceutisk "frigivelsestest." Frigivelsestest er et teknisk udtryk, der refererer til en proces, der involverer en række instrumentelle analyser, der bruges til omfattende testprodukter for renhed, koncentration, konsistens, identitet og urenheder af enhver art.
Hele FDA blev født af den ene "Department of Chemistry"-medarbejder fra 1862 og behovet for gennemsigtighed og verifikation af ingredienser. I dag har den medarbejder spredt sig til en hele FDA-afdelingen på 1,300 videnskabsmænd og støttepersonale formodet dedikeret til ingrediensverifikation via farmaceutisk frigivelsestest. FDA'erne Kontoret for farmaceutisk kvalitet (OPQ) formodes at sikre, at lægemidler nøjagtigt matcher indholdet af de anførte ingredienser, uden kvalitet/urenhed (kvalitativ) eller indholdsmæssig (kvalitativ) variabilitet. Reglerne, der kræver det, er meget specifikke og detaljerede i 21 CFR § 201.10.
Sådan verificerer FDA mRNA-injektioner til kvalitetskontrol:
Kvalitetskontrolresultaterne fra test fra mRNA-injektioner var særligt kritiske, fordi de er store, komplekse og blev hurtigt fremstillet. Mens skatteyderne er afhængige af FDA for at verificere mRNA-injektionskvaliteten og dele resultaterne, FDA synes forpligtet til at beskytte producenternes ingredienser på bekostning af selv den mest basale gennemsigtighed vedrørende mRNA Covid-produkter. Mens FDA ser ud til at indsamle prøver, er deres "mail-in"-metoden fundamentalt mangelfuld. Derudover deler FDA ikke resultaterne af disse tests nogen steder, hvor jeg kunne finde dem.
Med andre ord: under pandemien, da splinternye, bredt implementerede mRNA-skud blev skubbet over amerikanerne med "warp-hastighed", og da Amerika stolede mest på FDA's kvalitets-/regulative pligter, accepterede FDA selvindsendte "mail- i” kvalitetskontrol test og/eller resultater. Overvejede FDA det ikke mRNA-producenter indrømmede, at de "kæmper[d]" for at reagere på fremstillingen og "scramblede" for at følge med med fremstillingsprocesser? Producenter af mRNA-ingredienser udtalte endvidere, at bestræbelserne på at imødekomme behov var "uden fortilfælde."
Udsagn som denne giver ikke forbrugernes tillid til kvalitet og er illustrative for en enorm opskalering af disse komplekse produkter, som burde berettige især på vagt og personlig FDA-kontrol af faciliteter og fremstillede produkter, pandemi eller ej. En producent af mRNA-ingredienser sagde for eksempel, at de pludselig øgede deres produktion med 50 fold.
Midt i den nye teknologi, der blev presset igennem med "warp speed" hastværk, var der ingen af de 1,300 OPQ videnskabsmænd ved FDA, der krævede direkte inspektioner eller i det mindste tilbød at gøre andet end at bede om potentielt tvivlsomme "indsendte" prøver til test?
Det åbenlyse spørgsmål er: hvorfor indsamlede FDA ikke prøver direkte? Selv med pandemien på plads, kunne FDA have inspiceret faciliteter iført hazmat-dragter eller - eller på meget mindst – valgt at indsamle prøver fra apoteker, hospitaler eller på distributørers lagre.
Skjult metode til test af mRNA-injektionsingredienser:
Ud over fraværet af testresultater og tvivlsomme "indsendte" prøveudtagningsresultater - er FDA derudover skjuler deres validerede metodik, der forhindrer andre i at udføre deres egne, uafhængige analyser af kvaliteten/renheden af mRNA-injektioner.
Uafhængig analyse af lægemidler for renhed og potentiel kontaminering sammenlignet med ingredienslisten er noget, jeg selv havde forsøgt at gøre, da jeg konceptualiserede verdens første analytisk apotek. Men da mRNA-skud er en ny teknologi med en mindre end fuldt gennemsigtig ingrediensliste, er testmetoden, man skal bruge, ikke ligetil, som den ville være for andre småmolekylære lægemidler. Enhver, der forsøger at slå oplagringen, stabiliteten, specificiteten, kemien, sensitiviteten eller endda grundlæggende metodologi til at teste validering og/eller resultater, blokeres via en FDA-rapport, der indeholder latterligt invasive redaktioner, hvilket giver selv den mest fundamentale videnskabelige forståelse af, hvordan man potentielt kan evaluere resultater eller udføre test umuligt.
Som et gribende visuelt eksempel er en enkelt redigeret side i en længere FDA-regulativ oversigt (vist nedenfor) en del af en 127-sides dokument (hvoraf kun 63 sider er blevet delt, og af disse 63 sider er omkring 50 % blevet redigeret) om, hvordan man evaluerer renheden, koncentrationen og andre analytiske mål for mRNA-injektioner.
De FDA (b)(4) redaktioner specificerede detaljerede redaktioner brugt til "beskytter forretningshemmeligheder og fortrolige kommercielle eller finansielle oplysninger." Men er det virkelig passende at mærke det "kommercielt", hvis forskningen/udviklingen/produktet blev finansieret med hundreder af millioner af skatteydere?

Uden en liste over ingredienser eller testmetoder er det umuligt for andre uden for FDA eller producenter at vide præcist, hvordan man kontrollerer produktet forfalskning (ændrede eller giftige ingredienser) eller fejlmærkning (fordi en komplet liste over ingredienser inklusive nukleotidsekvensen og lipid nanopartikelkonfigurationer er særligt vage på produktetiketten).
Manglen på metodologi er særlig besværlig, da nye, foreløbige data ved brug af uafhængig metodologi har vist beviser for DNA-kontaminering i mRNA Covid-injektioner.
Så hvis en udefrakommende person hævdede at have testet og fundet en urenhed i mRNA-skud og bad FDA eller producenterne om dets svar, ville de blive mødt med et eller andet svar, der angiver noget i retning af:
- Du brugte ikke valideret/passende testmetode til at komme til dine konklusioner, og derfor er dine analyser ugyldige.
Til det vil det uafhængige laboratorium forsøge at anmode om testmetoden fra FDA-godkendt dokumentation (dvs. det fulde dokument, der indeholder Figur 5) ved at spørge: "Okay, jeg vil gerne teste det ved hjælp af din godkendte metode; vil du fortælle os, hvad det er?"
- FDA eller producenten ville svare noget i retning af: "Hvad vi er villige til at afsløre om den anvendte metode, som ikke er fortrolig, kan findes online eller via en FDA FOIA-anmodning” …hvor de ville blive mødt følgende stærkt redigerede dokument, hvor alt, der er yderst meningsfuldt, er dækket af (b)(4) redaktioner.
At læse mellem linjerne: Det er indlysende, at både producenter og amerikanske FDA ikke ønsker, at andre end dem selv skal kende de komplette ingredienser i eller endda teste mRNA-injektioner for renhed og konsistens.
Ifølge FDA embedsmænd: Pharmaceutical Manufacturing er Meget Udsat for fejl:
Mange ting kan – og gør – gå galt under den farmaceutiske fremstillingsproces. Ud over potentielle uoverensstemmelser med mRNA/LNP-injektioner implicerer kvalitative og kvantitative spørgsmål hver FDA-reguleret farmaceutisk produkt. Selv Parlamentet og Senatet har formelt anerkendt rapporter om FDA's manglende sikring af USA's farmaceutiske forsyningskæde. Størstedelen af Amerikas lægemiddel forbruger-slutbruger produkts bliver produceret i udlandet i lande som Indien og Kina, og andre lavlønomkostningslande er ikke velanset for høje niveauer af kvalitetskontrol. Det føderale register er fyldt med rapporter om krænkelser på indiske og kinesiske fabrikker.
Certificerer FDA også disse planter – inklusive dem med lang historie med overtrædelser – via et "mail-in" system til FDA? Skandaløst nok er svaret på spørgsmålet noget, der ville gøre enhver, der beskæftiger sig med farmaceutisk kvalitet, meget ubehagelig.
Mens en Six Sigma Præcisionsniveau har længe været målet for kvalitet og sikkerhed inden for bil-, computer-, mobiltelefon- og anden højteknologisk fremstilling, og det ser ud til at være blevet overset, når det kommer til farmaceutisk fremstilling.
FDA embedsmænd har offentliggjort data, der estimerer en upræcis på 2-3σ (sigma) i farmaceutisk fremstilling. En 2σ kvalitet svarer til 308,537 fejl pr. 1,000,000 muligheder. (Der er sandsynligvis meget mere end 1,000,000 muligheder for fejl, når det kommer til farmaceutisk fremstilling.) FDA er opmærksom på dette på de højeste niveauer af ledelse; faktisk strømmen FDA's leder af Office of Pharmaceutical Quality, Michael Kopcha selv skrev og publicerede ovenstående Six Sigma-beregning og beklagede den upræcise karakter af farmaceutisk fremstilling tilbage i 2017.
Fejlbredden for mRNA-produkter og/eller deres LNP'er kan være lige mindre præcise end 2-3σ, (jo lavere σ, jo mere fejlagtigt er et produkt), da de inkluderer nukleotidmateriale og nye LNP'er, hvilket gør dem væsentligt mere komplekse end småmolekylære lægemidler - til trods for at de er udviklet, fremstillet og frigivet på " kædehastighed."
Med selv FDA og dets embedsmænd anerkender en iboende produktionsunøjagtighed, hvorfor i sportens vide verden opfylder FDA ikke sin sikkerhedsmission ved offentligt at dele sin udgivelsestest af mRNA-teknologi med den amerikanske offentlighed, der finansierer dem?
Før 1862 igen? Er mRNA-skud de eneste stoffer, som amerikanere ikke har Komplet Ingrediensoplysninger?
Manglen på klarhed om antallet af sekvenser af mRNA-skud og anden kritisk information er i direkte kontrast til et andet FDA-godkendt RNA-baseret lægemiddel - patisiran (Onpattro®). Onpattro leverer gennemsigtigt sekvensen, molekylvægten og milligramstyrken af sine produkter inden for den officielle FDA emballagemærkning som illustreret i et uddrag nedenfor:


Mangel på Covid mRNA Dosisspecificitet: 0.3 ml (eller 0.5 ml) af hvad?
Lige nu har vi stadig ikke grundlæggende ingrediensoplysninger om nogen Covid mRNA-injektion. Farmaceuter ved kun at give en bestemt bind af væske, og tilsyneladende gjorde det uden spørgsmål. Normalt skal den officielle FDA-pakkemærkning detaljere de faktiske ingredienser i det volumen, men ikke for Covid mRNA-etiketter: de angiver blot 0.3 ml (eller 0.5 ml) som "Dosisform og styrke."
Derudover, som enhver gymnasieelev kan fortælle dig, er 0.3/0.5 ml en bind, ikke en styrke. Vi kender ikke nogen kvantitative detaljer om, hvad der er indeholdt i den 0.3/0.5 ml, såsom: Hvor mange LNP-partikler? Hvilken størrelse/morfologi af disse LNP'er? Hvor mange mRNA-sekvenser i det volumen?




Er dette, hvad der passer som tilstrækkeligt gennemsigtigt eller "sandfærdigt mærkning" af FDA?
Ovenstående klip-og-indsæt-uddrag fra indlægssedlen er alle de oplysninger, producenter deler med forbrugerne vedrørende dosis - som er sørgeligt utilstrækkelig sammenlignet med alle andre FDA-mærker - eller enhver, der er nysgerrig efter at vide noget ud over hvor meget væske at injicere og koncentrationen på 30 eller 100 mcg af en uspecificeret mRNA-sekvens.
Den bemærkelsesværdige unøjagtighed af denne etiket, der er tilladt af FDA, ser ud til at være i konflikt med dens næsten 120 år gamle etiket specifikt: "kræver, at fødevarer og lægemidler bærer sandfærdige mærkningserklæringer og opfylder visse standarder for renhed og styrke".
Er det det, der passer som en "sandfærdig" liste over ingredienser af FDA? (Se 21CFR §352og 21 CFR §201.10 vedrørende "angivelse af ingredienser" og "misbrandede lægemidler og enheder").
Spørgsmålet er: er der en liste over ukendte eller uspecifikke ingredienser, som ingen undtagen producenten kan tyde virkelig opfylder ånden eller lovkravene til "mærkning?" Er det mærke det, der anses for "sandfærdigt" af USA's FDA? Hvis side er FDA på alligevel; producenter eller forbrugere?
Ud over at det ikke er direkte specificeret, kan det nøjagtige antal af LNP- eller mRNA-strenge i en 30 eller 100 mcg injektion ikke engang ekstrapoleres støkiometrisk eller på baggrund af Avogadros nummer, fordi mRNA-sekvensen, molekylvægten og/eller LNP-komponenten/-konfigurationerne ikke er angivet nogen steder inden for den officielle FDA-mærkning.
Hvordan kan nogen vide, om antallet af mRNA-strenge til at kode for spikeproteinet for Covid er proportionalt med belastningen af Covid-podestof, som man ville modtage fra en samfundserhvervet infektion? Svar: de kan ikke.
Er Covid mRNA-injektioner Passende mærket/fejlmærket?
21 CFR 211.125 specificerer "Der skal udøves streng kontrol med mærkning, der er udstedt til brug ved mærkning af lægemidler,” men det ser ud til, at FDA var så slap med sin godkendte mærkning af Covid mRNA-injektioner på trods af at hvert andet lægemiddel - inklusive mRNA-baseret Onpattro - angiver denne information. Historisk set er FDA regulatoriske beslutninger (såsom hvilke oplysninger der skal inkluderes i produktmærkning) baseret på forrang, og Covid mRNA-skud var en åbenlys afvigelse fra FDA's historiske og juridiske forrang. Det bemærkelsesværdige datafravær og mangel på klarhed går på en måde tilbage til tiden Morleys lever- og nyrehjerte i slutningen af 1800-tallet. Forskellen er: dengang eksisterede FDA ikke, men i dag er der en FDA med ~20,000 ansatte, hvoraf i det mindste nogle tilsyneladende mente, at denne etiket var gennemsigtig og "sandfærdig".
At angive en ukendt/uoverskuelig/obskur ingrediens, som ingen nogensinde kunne fastslå nøjagtigt, er ikke, hvad lovgiverne fra 1906 Pure Food and Drug Act havde til hensigt, da de specificerede FDA-reglerne om "sandfærdig mærkning." Adskilt fra det: det faktum, at doserne fordobles pr. volumen fra forskellige producenter (30mcg/0.3mL vs 100mcg/0.5mL) betyder, at disse mRNA-sekvenser ser ud til at være vidt forskellige i nukleotidlængde og til gengæld ville have flere og forskellige LNP'er plus vedhæftede filer. Betyder det, at mRNA-sekvenser, der bruges til at transkribere spikeproteinet, er omkring dobbelt så store (10mcg/0.1mL versus 20mcg/0.1mL) sammenlignet med forskellige producenter, eller er der noget andet, der bidrager til nukleotidlængdeforskellen?
For lægmanden, der stadig læser indtil dette punkt (tillykke, forresten): Manglen på detaljerede mærkningsoplysninger kan være som at reklamere for et hus til salg, hvor det er lavet af træ og mursten på en cementplade – men ikke at vise. billeder af huset (f.eks. sekvens) og ikke deler dets kvadratoptagelser (f.eks. molekylvægt). Under alle omstændigheder er manglen på information utilstrækkelig og en afvigelse fra traditionelle standarder.
Hvert andet FDA-godkendt lægemiddel - inklusive andre mRNA-lægemidler - indeholder fuldstændige ingrediensoplysninger på deres produkter, inklusive en strukturel repræsentation og molekylvægt af deres produkt, så folk ved præcis, hvad de får.
Det er sandt: Slå op, hvad end du kan komme i tanke om i Drugs.com database og læg mærke til, hvordan alle mærker giver struktur og/eller molekylvægt. Bevis på, at Covid mRNA-skud er en iøjnefaldende undtagelse fra den historiske FDA-godkendelsespraksis og reglen om "sandfærdig etiket".
Danske undersøgelsesdetaljer 2023 Signifikant klinisk variation mellem batches af mRNA Covid-19 mRNA-injektioner:
Ikke at have nogen gennemsigtighed på selv potentielt ugyldige "indsendte" testvalidering ser ud til at have givet producenterne en videregivelse af en anden kritisk vigtig del af det, som FDA fører tilsyn med: potentielle kliniske manifestationer på parti-/batchvariationer af mRNA-skud. Et tilbageblik Dansk sikkerhedsundersøgelse offentliggjort tidligere i 2023 detaljerede et meget afvigende mønster af bivirkningsrapporter fra Pfizer-BioNTech BNT162b2 mRNA-injektioner som korreleret med det danske DKMA-bivirkningsrapporteringssystem.
I linjegrafen, der følger, repræsenterer forskellige farvede prikker forskellige partier af Pfizer-BioNTechs mRNA-injektioner. Det udskilte partier i tre forskellige kategorier; høj-lav- til (næsten) fraværende antal rapporterede bivirkningsgrupper (henholdsvis blå, grønne og gule plots).
Med andre ord: formodet "ækvivalente" produkter fra den samme producent ser ud til at have meget forskellige forekomster af uønskede hændelser, efter batch, hvor hver af disse batcher repræsenterer hundredtusindvis af mRNA-injektioner.
Når tilsvarende lineære regressionslinjer blev tilføjet, dukkede et bestemt mønster op:
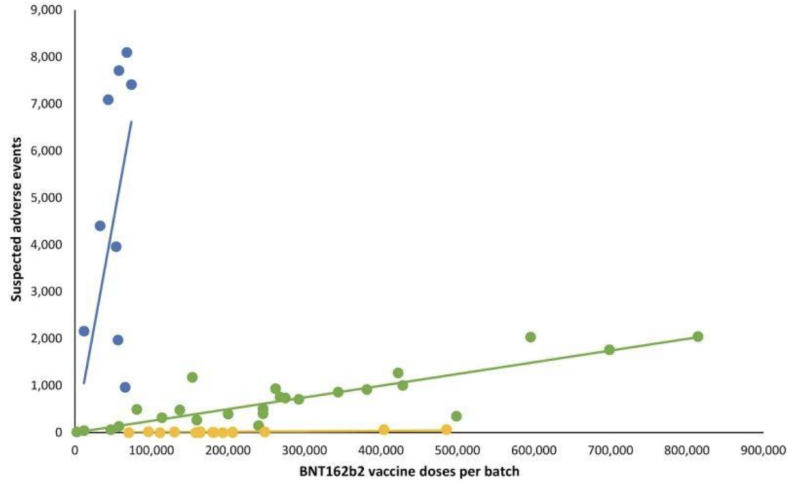
Vigtige spørgsmål om den bemærkelsesværdige uønskede hændelse forskel mellem Covid-19 mRNA batches omfatter:
- Kan uønskede hændelsesvarianser skyldes kvalitative eller kvantitative varianser i mRNA-sekvenser eller antallet af mRNA-strenge mellem batches?
- Kan uønskede hændelsesvarianser skyldes kvalitative eller kvantitative varianser i størrelsen/morfologierne eller mængden af LNP'er mellem batches? Hvilke test er blevet udført for sikre sikkerheden af forskellige LNP'er bruges i mRNA-injektioner?
- Var de batches, der svarede til de gule versus grønne versus blå datapunkter på en eller anden måde kvalitativt eller kvantitativt forskellige?
- Blev opbevaring/håndtering efter fremstilling kompromitteret på administrationsfaciliteten (eller et andet sted i forsyningskæden), hvilket førte til produktvariabilitet?
- Hvad er Sigma/fejlraten for dette og andre produkter, der stammer fra den særlige produktionsfacilitet/skiftechef med ansvar for fremstillingen?
- Blev ingredienser fra disse af Covid mRNA-produkter hentet fra Indien eller Kina i forhold til andre steder, afhængigt af batchen?
- Hvor mange procenter af partier af Covid mRNA-produkter blev testet via personlig indsamling af en FDA-inspektør i forhold til at blive "indsendt" fra start til dato? Blev hver enkelt batch testet ved kun at bruge en af disse to indsamlingsmetoder?
- Foretog FDA frigivelsestestverifikation på DKMA's partier til rapportering af uønskede hændelser? Hvis ja, hvorfor frigiver FDA ikke disse særlige testresultater? Hvis ikke, hvorfor blev der ikke testet?
- Er der et grundlæggende problem med konsekvent at producere LNP'er og/eller mRNA-sekvenser pålideligt og uden kontaminering?
Resultaterne af den danske undersøgelse og ovenstående spørgsmål om uønskede hændelser kunne *begynde* at blive behandlet, men ikke uden at FDA uafhængigt deler resultaterne af deres udgivelsestestresultater. Som det står, på grund af allestedsnærværende FDA (b)(4) redaktioner, kender ingen den validerede metode til at teste Covid mRNA-skud or præcis hvilke partier i den danske undersøgelse, der blev eller ikke blev testet or resultaterne af disse batchtests.
… Så igen, selvom FDA havde valgt at frigive disse batch-testresultater, hvordan ved forbrugerne, om disse resultater er repræsentative for de specificerede batches, da producenterne selv vælger, hvilke prøver der skal "mailes ind?"
Ikke at give ingrediensgennemsigtighed og sikre kvalitet via en passende prøveudtagningsmetode er et grundlæggende og grundlæggende krav fra FDA. Faktisk var det den primære årsag til dannelsen af FDA! Fortjener amerikanerne ikke bedre gennemsigtighed, tilsyn og "sandfærdig mærkning"-love, når det kommer til vores lægemidler - især da disse love blev lavet for over 100 år siden?
Udgivet under a Creative Commons Attribution 4.0 International licens
For genoptryk, sæt venligst det kanoniske link tilbage til originalen Brownstone Institute Artikel og forfatter.








