
In del et af denne artikel, gennemgik jeg de kontraktmæssige og regulatoriske rammer, som den amerikanske regering anvender til den indledende udvikling, fremstilling og erhvervelse af Covid mRNA-skud, ved at bruge BioNTech/Pfizer-aftalerne til at illustrere processen.
Jeg viste, at nødbrugstilladelse (EUA) blev givet til disse produkter baseret på kliniske forsøg og fremstillingsprocesser udført med
- ingen bindende juridiske standarder,
- ingen lovligt forbudt sikkerhedstilsyn eller regulering, og
- ingen juridisk erstatning fra producenten for potentielle skader.
I denne opfølgende artikel vil jeg give en detaljeret analyse af den bagvedliggende dokumentation.
Anden transaktionsmyndighed/-aftale (OTA): En Military Acquisition Pathway
aftale mellem den amerikanske regering, repræsenteret ved Department of Defense (DoD), og Pfizer, der repræsenterer BioNTech/Pfizer-partnerskabet, i juli 2020, for køb af en "vaccine til forebyggelse af COVID-19" var ikke en almindelig opkøbskontrakt.
Det var en aftale under Other Transaction Authority (OTA) – en opkøbsvej, der iflg Retningslinjer for forsvarsministeriet, er blevet brugt siden 1958 til at "tillade et føderalt agentur at indgå andre transaktioner end kontrakter, tilskud eller samarbejdsaftaler".
[BOLDANSIGT TILFØJET]
En grundig gennemgang af DoD's brug af OTA, herunder dets lovpligtige historie, kan findes i 22. februar 2019 Congressional Research Service-rapport. Denne rapport specificerer sammen med enhver anden diskussion af OTA, at det er en alternativ opkøbsvej til forsvar og militære formål. Det er ikke beregnet til, og har heller aldrig været brugt før Covid, til noget, der primært er beregnet til civilt brug.
Hvis du leder efter OTA-love i US Code, dette er den vej, du vil gå ned ad:
Væbnede styrker -> Generel militærlovgivning -> Erhvervelse -> Forskning og teknik -> Aftaler -> DoDs myndighed til at udføre visse prototypeprojekter
Denne juridiske vej viser meget tydeligt, at OTA-love er beregnet til erhvervelse af forsknings- og ingeniørprototyper til de væbnede styrker.
Forsvarsministeriet har autoritet til tre forskellige typer OT'er: (1) forsknings-OT'er, (2) prototype-OT'er og (3) produktions-OT'er.
Disse tre typer OT'er repræsenterer tre stadier af indledende forskning, udvikling af en prototype og eventuel produktion.
Inden for disse tre typer er der specifikke kategorier af projekter, som OTA kan ansøge om:
- Oprindeligt ifølge OTA Oversigt leveret af DoD, var den anden transaktionsmyndighed "begrænset til at gælde for våben eller våbensystemer, der foreslås erhvervet eller udviklet af DoD."
- OTA blev senere udvidet til at omfatte "ethvert prototypeprojekt, der er direkte relateret til at forbedre missionseffektiviteten af militært personel og de understøttende platforme, systemer, komponenter eller materialer, der foreslås erhvervet eller udviklet af DoD, eller til forbedring af platforme, systemer, komponenter , eller materialer i brug af Forsvaret."
Indtil videre lyder intet af det som en anskaffelsesvej for millioner af nye medicinske produkter, der primært er beregnet til civilt brug.
Er der nogen undtagelse for civil brug af OTA, der kan gælde for Covid mRNA-vacciner?
FY2004 National Defense Authorization Act (PL 108-136) indeholdt et afsnit, der gav Anden Transaktionsmyndighed til "lederen af et forvaltningsorgan, der beskæftiger sig med grundforskning, anvendt forskning, avanceret forskning og udviklingsprojekter", der "har potentialet til at lette forsvar mod eller genopretning fra terrorisme eller nuklear, biologisk, kemisk eller radiologisk angreb."
Denne bestemmelse blev forlænget indtil 2018, men ser ikke ud til at være forlænget ud over dette år. Bemærk også, at selv i dette ekstraordinære tilfælde af ikke-DoD brug af OTA, situationen skal involvere terrorisme eller et angreb med masseødelæggelsesvåben (CBRN).
Hvilke andre OTA-love kan være gældende?
2019 CRS-rapporten citeret ovenfor giver dette diagram, der viser, at nogle få ikke-DoD-agenturer har nogle OTA eller relaterede myndigheder:
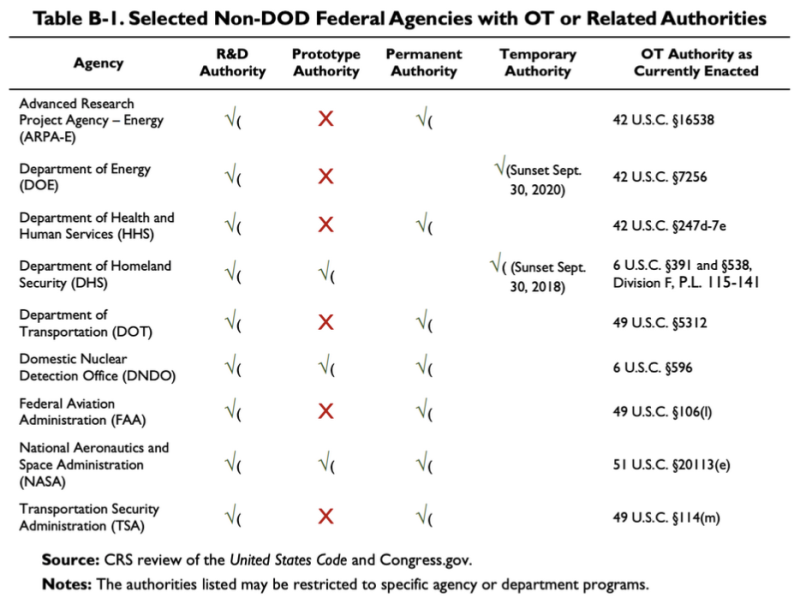
Ifølge denne tabel har Department of Health and Human Services (HHS) nogle andre transaktionsmyndigheder for forskning og udvikling (F&U). Loven vedr OT Authority of HHS er 42 U.S.C. §247d-7e.
Hvor er denne lov placeret, og hvad siger den?
The Public Health and Welfare -> Public Health Service -> Generelle beføjelser og pligter -> Federal-State Cooperation -> Biomedical Advanced Research and Development Authority (BARDA) -> Transaktionsmyndigheder
Så der er et sted i loven relateret til civil sundhed og velfærd, hvor OTA kan være gældende, selvom den er gyldig kun til forskning og udvikling, ikke prototyper eller fremstilling.
Loven siger, at BARDA-sekretæren har OT-autoritet
med hensyn til et produkt, der er eller kan blive en kvalificeret modforanstaltning eller kvalificeret pandemi eller epidemisk produkt, aktiviteter, der overvejende—
(i) udføres efter grundforskning og præklinisk udvikling af produktet og
(ii) er relateret til fremstilling af produktet i kommerciel skala og i en form, der opfylder de lovmæssige krav under Federal Lov om fødevarer, stoffer og kosmetik [21 USC 301 et seq.] eller under afsnit 262 i denne titel.
[BOLDANSIGT TILFØJET]
De "lovgivningsmæssige krav", der er opregnet i loven, betyder, at det ville være umuligt for BARDA/HHS at indgå aftaler – selv bare F&U – for medicinske produkter (som mRNA-vaccinerne), der ikke har gennemgået streng sikkerhedstest og strengt produktionstilsyn.
HHS "Partnerskab" med DoD omgået civilbeskyttelseslove
For at opsummere den vanskelige situation med andre transaktionsmyndigheder/-aftaler med hensyn til civile myndigheder i almindelighed og Covid mRNA-vacciner, i særdeleshed:
- OTA blev skrevet og kodificeret som en måde for militæret at erhverve våben og andre nødvendige systemer og udstyr uden en masse bureaukratisk bureaukratisk. Det dækker forskning og udvikling, prototyper og efterfølgende fremstilling.
- Den eneste OTA for et folkesundhedsagentur er for HHS, og det dækker kun forskning og udvikling, ikke prototyper eller fremstilling.
- Selv R&D OTA givet til HHS kræver stadig, at produkter fremstilles "i en form, der opfylder de lovmæssige krav" til lægemiddel- og vaccinesikkerhed.
Med andre ord: Der er ingen måde, HHS kunne have brugt sin meget begrænsede OTA til at underskrive kontrakter for hundreder af millioner af nye medicinske produkter.
Så hvad gjorde HHS?
Som Government Accountability Office (GAO) bemærkede i sin Juli 2021-rapport om "Covid-19-kontrakter:" HHS "samarbejdede" med DoD for at "udnytte DoD's OTA-myndigheder ... som HHS manglede." (s. 24)
Hvad er DoDs OT-myndigheder for medicinske produkter?
Som diskuteret er OTA beregnet til at hjælpe militæret med at få udstyr og teknologi uden masser af bureaukratisk besvær. Ingen af de oprindelige love vedrørende OTA nævnte andet end "platforme, systemer, komponenter eller materialer" beregnet til at "forbedre missionseffektiviteten for militært personel."
Men fem år før Covid blev der indført en usædvanlig brug af OTA:
I 2015, blev DoD annonceret etableringen af CBRN Medical Countermeasure Consortium, hvis formål var at bruge OTA-opkøbsvejen til at "arbejde sammen med DoD om at udvikle FDA-licenserede kemiske, biologiske, radiologiske og nuklearmedicinske modforanstaltninger." [FDA = Food & Drug Administration]
Som beskrevet i 2015-meddelelsen inkluderede dette "prototypeteknologier til terapeutiske medicinske modforanstaltninger rettet mod virale, bakterielle og biologiske toksinmål af interesse for DoD." Listen over midler omfattede de bedste biowarfare-patogener, såsom miltbrand, ebola og marburg.
Meddelelsen fortsatte med at specificere, at "aktiverende teknologier kan omfatte dyremodeller af viral, bakteriel eller biologisk toksinsygdom og patogenese (flere eksponeringsveje), assays, diagnostiske teknologier eller andre platformsteknologier, der kan anvendes til udvikling af godkendte eller licenserede MCM'er [medicinske modforanstaltninger]."
Selvom dette stadig ikke lyder som produktionen af 100 millioner nye vacciner til civil brug, giver det mere spillerum for OTA end den meget begrænsede Other Transaction Authority givet til HHS.
Mens HHS OTA kræver overholdelse af omfattende udviklings- og fremstillingsregler, kræver OTA-vejen for DoD til at udvikle medicinske modforanstaltninger kun "FDA-licens."
Ved hjælp af DoD Other Transaction Authorities vil det således teoretisk være muligt at omgå alle sikkerhedsbestemmelser – afhængigt af kravene til FDA-licensering af et OTA-genereret produkt. Som vi vil se, blev der i tilfælde af Covid mRNA-vacciner givet nødbrugstilladelse, hvilket overhovedet ikke kræver juridisk sikkerhedstilsyn.
Tilladelse til nødbrug (EUA)
Her er hvordan Food & Drug Administration (FDA) beskriver sine EUA-beføjelser:
Section 564 of the FD&C Act (21 USC 360bbb – 3) giver FDA mulighed for at styrke beskyttelsen af folkesundheden mod biologiske, kemiske, nukleare og radiologiske agenser.
Med denne EUA-autoritet kan FDA hjælpe med at sikre, at medicinske modforanstaltninger kan bruges i nødstilfælde til at diagnosticere, behandle eller forebygge alvorlige eller livstruende sygdomme eller tilstande forårsaget af biologiske, kemiske, nukleare eller radiologiske agenser, når der ikke er tilstrækkelige, godkendte , og tilgængelige alternativer (blandt andre kriterier).
Det er ekstremt vigtigt at forstå, at disse EUA-beføjelser blev givet i 2004 under meget specifikke omstændigheder relateret til beredskab til angreb med masseødelæggelsesvåben, ellers kendt som CBRN-midler (kemiske, biologiske, radiologiske, nukleare).
Som forklaret i Harvard Law's Bill of Health,
I sidste ende var det War on Terror, der ville give anledning til nødbrugstilladelse. Efter begivenhederne den 11. september 2001 og efterfølgende miltbrandpostangreb vedtog kongressen Project Bioshield Act af 2004. Loven krævede milliarder af dollars i bevillinger til køb af vacciner som forberedelse til et bioterrorangreb og til oplagring af nødforanstaltninger. For at kunne handle hurtigt i en nødsituation tillod kongressen FDA at godkende formelt ikke-godkendte produkter til brug i nødstilfælde mod en trussel mod folkesundheden og sikkerheden (med forbehold for en nøderklæring fra HHS). Det optage indikerer, at kongressen var fokuseret på truslen om bioterror specifikt, ikke på at forberede sig på en naturligt forekommende pandemi.
ordlyden af EUA-loven understreger, at det var beregnet til brug i situationer, der involverede masseødelæggelsesvåben. Her er de 4 situationer, hvor EUA kan udstedes:
- en afgørelse fra Secretary of Homeland Security om, at der er en indenlandsk nødsituation eller et betydeligt potentiale for en indenlandsk nødsituation, der involverer en øget risiko for angreb med et eller flere biologiske, kemiske, radiologiske eller nukleare agenser;
- en afgørelse fra forsvarsministeren om, at der er en militær nødsituation eller et betydeligt potentiale for en militær nødsituation, der involverer en øget risiko for United Stater militære styrker, herunder personel, der opererer under myndigheden i afsnit 10 eller afsnit 50, angreb med—
- et eller flere biologiske, kemiske, radiologiske eller nukleare agenser; eller
- en agent eller agenter, der kan forårsage eller på anden måde er forbundet med en overhængende livstruende og specifik risiko for United Stater militære styrker;
- en bestemmelse af Sekretær at der er en folkesundhedsnødsituation eller et betydeligt potentiale for en folkesundhedsnødsituation, der påvirker eller har et betydeligt potentiale til at påvirke den nationale sikkerhed eller Uniteds sundhed og sikkerhed Stater borgere, der bor i udlandet, og som involverer et eller flere biologiske, kemiske, radiologiske eller nukleare agenser eller en sygdom eller tilstand, der kan tilskrives sådanne agenser; eller
- identifikation af en væsentlig trussel i henhold til § 319F-2 i Lov om offentlig sundhedstjeneste [42 USC 247d–6b] tilstrækkelig til at påvirke den nationale sikkerhed eller Uniteds sundhed og sikkerhed Stater borgere, der bor i udlandet.
Ingen steder i disse fire situationer er der nogen omtale af en naturligt forekommende epidemi, pandemi eller nogen anden form for folkesundhedssituation, der ikke er forårsaget af "biologiske, kemiske, radiologiske eller nukleare agenser."
Kunne SARS-CoV-2 kvalificere sig som en sådan agent?
Hvis du leder efter definitionen af "biologiske midler” i US Legal Code, vil du gå ned ad følgende vej:
Forbrydelser og straffesager -> Forbrydelser -> Biologiske våben -> Definitioner
Så i sammenhæng med amerikansk lovgivning betyder udtrykket "biologiske midler" biologiske våben, og brugen af sådanne midler/våben betragtes som en forbrydelse.
Wikipedia giver dette definition:
Et biologisk middel (også kaldet bio-agent, biologisk trusselmiddel, biologisk krigsførelsesmiddel, biologisk våben eller biovåben) er et bakterie, virus, protozo, parasit, svamp, eller toksin, der kan bruges målrettet som et våben i bioterrorisme or biologisk krigsførelse (BW).
På hvilket juridisk grundlag blev EUA udstedt for Covid mRNA-vacciner?
Det ser ud til, baseret på lovene vedrørende EUA, at ingen af de fire mulige situationer beskrevet i loven kunne anvendes på et produkt, der er beregnet til at forebygge eller behandle en sygdom forårsaget af et naturligt forekommende patogen.
Ikke desto mindre blev denne lov brugt til at godkende mRNA Covid-vaccinerne.
I betragtning af de fire valg, der er anført i EUA-loven, var den, der blev brugt til Covid-"modforanstaltninger",
C) en bestemmelse af Sekretær at der er en folkesundhedsnødsituation eller et betydeligt potentiale for en folkesundhedsnødsituation, der påvirker eller har et betydeligt potentiale til at påvirke den nationale sikkerhed eller Uniteds sundhed og sikkerhed Stater borgere, der bor i udlandet, og som involverer et eller flere biologiske, kemiske, radiologiske eller nukleare agenser eller en sygdom eller tilstand, der kan tilskrives sådanne agenser.
Hvornår anvendes specifikt til Covid, sådan blev det formuleret:
sekretæren for Department of Health and Human Services (HHS) fastslog, at der er en folkesundhedsnødsituation, der har et betydeligt potentiale til at påvirke den nationale sikkerhed eller sundheden og sikkerheden for amerikanske borgere, der bor i udlandet, og som involverer den virus, der forårsager Coronavirus Sygdom 2019 (COVID-19)...
Der er her ingen tvivl om, at "virussen, der forårsager COVID-19" anses for at være ækvivalent med "et biologisk, kemisk, radiologisk eller nukleart agens eller midler."
Det er også vigtigt at bemærke, at EUA's "bestemmelse af en nødsituation for folkesundheden" er fuldstændig adskilt fra og ikke på nogen måde afhængig af andre nøderklæringer om folkesundhed, som dem, der blev afgivet af WHO, den amerikanske regering , og præsidenten i begyndelsen af Covid-19-pandemien.
Så selv når WHO, den amerikanske regering og præsidenten erklærer, at pandemien er forbi, kan der stadig være tilladelse til brug i nødstilfælde, hvis HHS-sekretæren fortsætter med at hævde, at situationen beskrevet i afsnit C) eksisterer.
Ser man på alle EUA'er for hundredvis af Covid-relaterede medicinske produkter, er det meget svært at se, hvordan HHS-sekretæren kunne retfærdiggøre påstanden om, at "der er en folkesundhedsnødsituation, der har et betydeligt potentiale til at påvirke den nationale sikkerhed eller sundheden og sikkerheden for amerikanske borgere, der bor i udlandet" i de fleste, hvis ikke alle, af disse sager.
Yderligere "lovpligtige kriterier" for FDA til at give tilladelse til brug i nødstilfælde
Når først HHS-sekretæren erklærer, at der er en nødsituation for folkesundheden, der berettiger EUA, baseret på en af de fire situationer, der er anført i loven, er der yderligere fire "lovpligtige kriterier", der skal opfyldes, for at FDA kan udstede EUA . Her er, hvordan FDA forklarer disse krav:
- Alvorlig eller livstruende sygdom eller tilstand
For at FDA kan udstede en EUA, skal CBRN-agenserne, der henvises til i HHS-sekretærens EUA-erklæring, være i stand til at forårsage en alvorlig eller livstruende sygdom eller tilstand.
BEMÆRK: Dette kriterium gentager specifikationen af en CBRN-agent, som er juridisk defineret som et våben, der bruges til at begå en forbrydelse.
- Bevis på effektivitet
Medicinske produkter, der kan komme i betragtning til en EUA, er dem, der "kan være effektive" til at forebygge, diagnosticere eller behandle alvorlige eller livstruende sygdomme eller tilstande, der kan være forårsaget af en eller flere CBRN-agenser identificeret i HHS-sekretærens erklæring om nødsituation eller trussel om nødsituation i henhold til paragraf 564(b).
Standarden "kan være effektiv" for EUA'er giver et lavere bevisniveau end den "effektivitet"-standard, som FDA bruger til produktgodkendelser. FDA har til hensigt at vurdere den potentielle effektivitet af et muligt EUA-produkt fra sag til sag ved hjælp af en risiko-benefit-analyse, som forklaret nedenfor.
[BOLDANSIGT TILFØJET]
JURIDISK SPØRGSMÅL: Hvordan kan nogen lovligt hævde, at et produkt, der er godkendt i henhold til EUA, er "sikkert og effektivt", hvis den juridiske standard for EUA er "kan være effektiv", og FDA erklærer, at dette er et "lavere niveau af bevis" end den anvendte standard for almindelige produktgodkendelser?
- Risiko-Benefit Analyse
Et produkt kan komme i betragtning til en EUA, hvis kommissæren fastslår, at de kendte og potentielle fordele ved produktet, når det bruges til at diagnosticere, forebygge eller behandle den identificerede sygdom eller tilstand, opvejer de kendte og potentielle risici ved produktet.
Ved at afgøre, om de kendte og potentielle fordele ved produktet opvejer de kendte og potentielle risici, har FDA har tænkt sig at kigge på det samlede videnskabelige bevis for at foretage en overordnet risiko-benefit-bestemmelse. Sådanne beviser, som kunne opstå fra forskellige kilder, kan omfatte (men er ikke begrænset til): resultater af indenlandske og udenlandske kliniske forsøg, in vivo-effektivitetsdata fra dyremodeller og in vitro-data, tilgængelig for FDA-betragtning. FDA vil også vurdere kvaliteten og kvantiteten af tilgængelige beviseri betragtning af den nuværende videnskabelige viden.
[BOLDANSIGT TILFØJET]
JURIDISK NOTE: Der er ingen juridisk standard, og der er ingen juridiske definitioner for, hvad det betyder, at "kendte og potentielle fordele" opvejer "kendte og potentielle risici." Der er heller ingen kvalitativ eller kvantitativ juridisk definition af, hvad der udgør acceptabel "tilgængelig dokumentation", som risiko-benefit-analysen "kan være" baseret på. Der kunne være nul faktisk bevis, men en tro på, at et produkt har en masse potentielle fordele og ikke en masse potentielle risici, og det ville opfylde dette "lovpligtige krav."
- Ingen alternativer
For at FDA kan udstede en EUA, må der ikke være noget passende, godkendt og tilgængeligt alternativ til kandidatproduktet til diagnosticering, forebyggelse eller behandling af sygdommen eller tilstanden. Et potentielt alternativt produkt kan betragtes som "utilgængeligt", hvis der ikke er tilstrækkelige forsyninger af det godkendte alternativ til fuldt ud at opfylde nødbehovet.
JURIDISK FORESPØRGSEL: Bortset fra den grove og potentielt kriminelle bagvaskelse/forbud mod alternative Covid-19-behandlinger som ivermectin og hydroxychloroquin, på hvilket tidspunkt var der et godkendt alternativ til at "forebygge Covid-19" (det eneste mRNA-vaccinerne blev købt for at gøre ) – Paxlovid, for eksempel – hvilket ville gøre en EUA for mRNA-vaccinerne ikke længere lovlig?
Her er hvordan alle disse "lovpligtige kriterier" blev opfyldt i virkeligheden Nødbrugstilladelse for BioNTech/Pfizer Covid mRNA-vaccinerne:
Jeg har konkluderet, at nødanvendelse af Pfizer-BioNTech COVID-19-vaccine til forebyggelse af COVID-19, når den administreres som beskrevet i scope of Authorization (Section II) opfylder kriterierne for udstedelse af en autorisation i henhold til Section 564(c) af loven, fordi:
- SARS-CoV-2 kan forårsage en alvorlig eller livstruende sygdom eller tilstand, herunder alvorlig luftvejssygdom, hos mennesker inficeret med denne virus;
- Baseret på alle de videnskabelige beviser, der er tilgængelige for FDA, er det rimeligt at tro, at Pfizer-BioNTech COVID-19 Vaccine kan være effektiv til at forebygge COVID-19, og at de kendte og potentielle fordele ved Pfizer-BioNTech COVID-19 Vaccine, når de anvendes under de betingelser, der er beskrevet i denne godkendelse når det bruges til at forebygge COVID-19 opveje dets kendte og potentielle risici; og
- Der er ikke noget passende, godkendt og tilgængeligt alternativ til nødbrug af Pfizer-BioNTech COVID-19-vaccinen for at forebygge COVID-19.
[BOLDANSIGT TILFØJET]
BEMÆRK: Den eneste kontekst, hvor FDA afvejede de potentielle fordele og risici ved vaccinen, og hvor FDA fastslog, at den "kan være effektiv", var med at forebygge Covid-19.
Der er ingen overvejelser, ingen beviser for faktisk eller potentiel fordel, og ingen afgørelse om, at der er nogen potentiel effektivitet for vaccinen til at gøre noget andet, herunder: sænke risikoen for alvorlig sygdom, sænke risikoen for hospitalsindlæggelse, sænke risikoen for død , hvilket sænker risikoen for forhold, der faktisk eller potentielt er relateret til Covid-19.
DERFOR kan man med rimelighed stille spørgsmålstegn ved lovligheden af enhver påstand om, at vaccinen er "sikker og effektiv" i forbindelse med alt andet end "når den bruges til at forhindre COVID-19" - hvilket man vidste, at vaccinerne IKKE GØR meget kort tid efter, at de blev indført.
Hvis folk fik at vide, at BioNTech/Pfizer mRNA-vaccinerne var "sikre og effektive" til andet end at forhindre Covid-19, og hvis de blev truet med konsekvenser for manglende vaccination til andet end at forhindre Covid-19, kunne de måske har et legitimt argument for, at de ulovligt blev tvunget til at tage et ikke-godkendt produkt under svigagtige krav?
Tredje niveaus krav til EUA for ikke-godkendte produkter
Når vi først har den EUA-specifikke nøddeklaration, og når FDA erklærer, at produktet kan være effektivt, og at alt det bevis, der er tilgængeligt (fra nul til uendeligt), viser, at dets fordele opvejer dets risici (som bestemt af hvad FDA mener, at de kan være), er der endnu et lag af ikke-sikkerhedsmæssig, ikke-effektivitetsrelateret regulering.
Her er hvordan en 2018 Congressional Research Service rapport om EUA forklarer dette:
FFDCA §564 pålægger FDA at pålægge visse påkrævede betingelser i en EUA og giver mulighed for yderligere skønsmæssige betingelser, hvor det er relevant. De påkrævede betingelser varierer afhængigt af, om EUA er for et ikke-godkendt produkt eller for en ikke-godkendt brug af et godkendt produkt. For et ikke-godkendt produkt skal brugsbetingelserne:
(1) sikre, at sundhedspersonale, der administrerer produktet, modtager den nødvendige information;
(2) sikre, at personer, som produktet administreres til, modtager den nødvendige information;
(3) sørge for overvågning og rapportering af uønskede hændelser forbundet med produktet; og
(4) sørge for registrering og rapportering fra fabrikantens side.
JURIDISK SPØRGSMÅL: Hvad er egentlig de "påkrævede oplysninger?" Vi ved, at folk blev informeret om, at vaccinerne fik nødbrugstilladelse. Men fik de at vide, at dette betyder "et lavere niveau af bevis" end det, der kræves for "sikre og effektive" påstande om andre medicinske produkter? Blev de informeret om, at der er forskellige niveauer af "sikker og effektiv" afhængig af, om et produkt har EUA eller en anden type godkendelse?
BEMÆRK: Loven kræver, at der er en måde at overvåge og rapportere uønskede hændelser på. Det fremgår dog ikke, hvem der overvåger, hvilke standarder der er for rapportering, og hvad tærsklen er for at handle på baggrund af rapporterne.
EUA sammenlignet med alle andre lægemidler/vacciner godkendelsesvej
Som forsker/skribent Sasha Latypova har påpeget, at mange mennesker blev forvirrede af EUA, fordi det lyder meget som EAU, som står for "Expanded Access Use." Dette er en type godkendelse, der gives til medicinske produkter, når der er et presserende behov for en bestemt gruppe patienter (f.eks. kræftpatienter i fase IV, hvis forventede levetid måles i måneder), som er villige til at risikere uønskede hændelser og endda døden i bytte for adgang til en eksperimentel behandling.
Nødbrugsautorisation er på ingen måde relateret til, og har heller ingen lighed med, udvidet adgangsbrug.
De forskellige juridiske veje til godkendelse af medicinske produkter er pænt præsenteret i en tabel fremhævet af juridisk forsker Katherine Watt. Tabellen er en del af en 2020-præsentation for en FDA-CDC Joint Learning Session: Regulatoriske opdateringer om brug af medicinske modforanstaltninger.
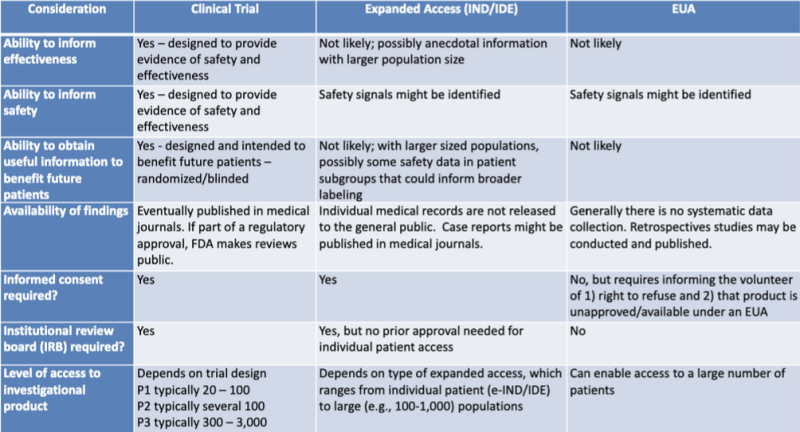
Denne tabel viser meget tydeligt, at EUA-processen sandsynligvis ikke giver information om produktets effektivitet, ikke er designet til at give bevis for sikkerhed, sandsynligvis ikke vil give nyttige oplysninger til gavn for fremtidige patienter, involverer ingen systematisk dataindsamling, kræver ingen retrospektive undersøgelser, intet informeret samtykke og intet institutionelt bedømmelsesudvalg.
Desuden i en 2009 Institute of Medicine af National Academic publikation, også fremhævet af Watt, med titlen "Medical Countermeasures: Dispensing Emergency Use Authorization and the Postal Model - Workshop Summary" finder vi denne erklæring på s. 28:
Det er vigtigt at erkende, at en EUA ikke er en del af udviklingsforløbet; det er en helt separat enhed, der kun bruges i nødsituationer og er ikke en del af lægemiddelgodkendelsesprocessen.
Betyder det, at godkendelser af Covid-19 modforanstaltninger, der var baseret på EUA'er, var ulovlige? Betyder det, at der ikke er nogen lovlig måde at hævde, at et EUA-produkt er "sikkert og effektivt", fordi det IKKE er EN DEL AF LÆGEMIDDELGODKENDELSESPROCESSEN?
Konklusion
Det er yderst tydeligt, givet alle oplysningerne i denne artikel og i den foregående del 1, at BioNTach/Pfizer Covid mRNA-vaccinerne blev udviklet, fremstillet og godkendt i henhold til militærlovgivning forbeholdt nødsituationer, der involverer biologisk krigsførelse/terrorisme, ikke naturligt forekommende sygdomme, der påvirker hele civilbefolkningen.
Derfor var overholdelse af regler og tilsyn, som vi forventer at finde, når et produkt anses for "sikkert og effektivt" for hele civilbefolkningen, ikke lovpligtigt.
Kan denne analyse bruges til at anfægte lovligheden af det "sikre og effektive" krav fra de embedsmænd, der vidste, hvad EUA indebar? Er der andre juridiske konsekvenser?
Det håber jeg.
Vigtigere er det, at i juridiske udfordringer til Covid mRNA-vacciner, der er bragt indtil videre, har der ikke været nogen afgørelser (som jeg er bekendt med) om hvorvidt militærlovgivning, som OTA og EUA, kan anvendes på civile situationer. Der har dog været en udtalelse fra byretsdommer Michael Truncale, i sin afvisning af sagen om whistleblower Brook Jackson mod Ventavia og Pfizer, det er vigtigt at huske på.
Her anerkender dommeren, at aftalen om BioNTech/Pfizer mRNA-vaccinerne var en militær OTA, men han nægter at tage stilling til dens anvendelighed på de ikke-militære omstændigheder (naturligt forekommende sygdom, 100 millioner doser, for det meste ikke til militær brug), hvorunder den blev udstedt:
Det faktum, at både militært personel og civile modtog vaccinen, indikerer ikke, at erhvervelsen af vaccinen var irrelevant for at forbedre militærets missionseffektivitet. Endnu vigtigere er det, at fru Jackson i virkeligheden beder denne domstol om at tilsidesætte DoD's beslutning om at udøve anden transaktionsmyndighed til at købe Pfizers vaccine. Men som USA's højesteret længe har understreget, er de "komplekse subtile og professionelle beslutninger med hensyn til sammensætning, træning, udrustning og kontrol af en militær styrke i det væsentlige professionelle militære domme." Gilligan v. Morgan, 413 US 1, 10 (1973). Det er således "svært at forestille sig et område med statslig aktivitet, hvor domstolene har mindre kompetence." Id. Denne domstol vil ikke nedlægge veto mod DoD's domme vedrørende missionens effektivitet under en national nødsituation.
Dette er blot en af mange juridiske forhindringer, der er tilbage i kampen for i sidste ende at forbyde alle mRNA-produkter, der er godkendt under Covid-19-nødsituationen, og alle efterfølgende mRNA-produkter, hvis godkendelse var baseret på Covid-19-godkendelsesprocessen.
Udgivet under a Creative Commons Attribution 4.0 International licens
For genoptryk, sæt venligst det kanoniske link tilbage til originalen Brownstone Institute Artikel og forfatter.








