
Under Covid-pandemien brugte den amerikanske regering milliarder af dollars på næsten 400 produkter beregnet til at beskytte, diagnosticere og behandle hundredvis af millioner af mennesker - alle med mærket "EUA" eller "Emergency Use Authorization."
Men hvad betyder EUA egentlig?
Allerede før vi besvarer det spørgsmål, og for at forstå, hvor EUA står i forhold til andre veje til at godkende eller godkende medicinske produkter, er det nyttigt at se på hvad EUA ikke er:
EUA er ikke en betegnelse for et eksperimentelt produkt, der gennemgår et klinisk forsøg
Hvis vi kun forstår én ting om EUA, burde det være dette: EUA gælder ikke for et produkt, der gennemgår et klinisk forsøg, der er underlagt FDA-bestemmelser eller andre lovkrav.
EUA er heller ikke det samme som Expanded Access Use (EAU), ofte kaldet "compassionate use"-adgang, som gælder for at give patienter med svære, uhelbredelige sygdomme adgang til eksperimentelle produkter, før de er fuldt godkendte.
Dette bord fra en FDA-CDC 2020 præsentation opsummerer forskellene mellem produkter, der gennemgår kliniske forsøg, produkter givet til patienter gennem udvidet "medfølende" adgang og produkter godkendt gennem EUA:
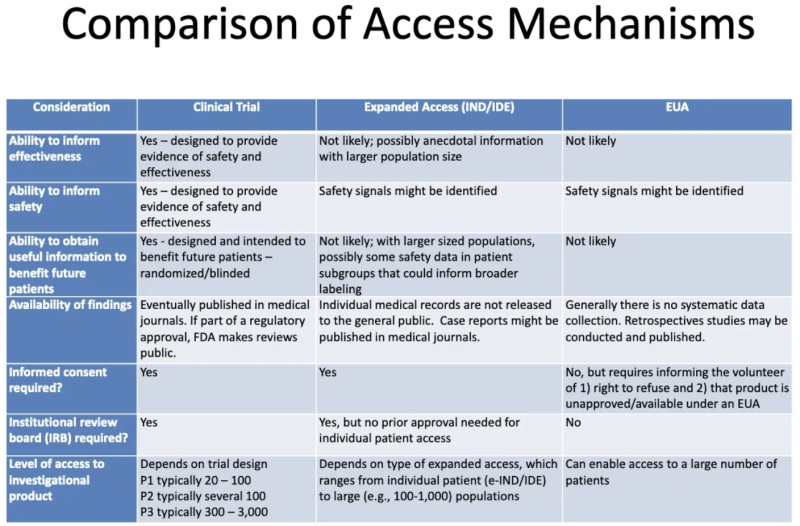
Her er, hvad denne tabel fortæller os om EUA:
- Processen med at tildele EUA vil sandsynligvis ikke generere nogen information om et produkts effektivitet.
- Processen med at tildele EUA er ikke designet til at give bevis for sikkerhed eller effektivitet, men sikkerhedssignaler kan identificeres.
- Det er usandsynligt, at når et produkt først er tildelt EUA og administreret til nogle patienter, vil der blive indhentet brugbar information til gavn for fremtidige patienter.
- Der er ingen systematisk dataindsamling om effektivitet eller sikkerhed med EUA, og ingen data offentliggøres i medicinske tidsskrifter som en del af den regulatoriske godkendelsesproces.
- Der kræves intet informeret samtykke, men patienter, der " melder sig frivilligt " til at tage produktet, skal få at vide, at de kan afslå, og at produktet ikke er godkendt/tilgængeligt under EUA.
- Der kræves ikke noget institutionelt revisionsudvalg (IRB). [IRB er en bestyrelse, der formodes at beskytte menneskers velbefindende i kliniske forsøg]
For at præcisere endnu mere, hvor adskilt EUA er fra enhver normal godkendelsesproces, i en 2009 Institute of Medicine of the National Academies publikation, finder vi denne erklæring:
Det er vigtigt at erkende, at en EUA ikke er en del af udviklingsforløbet; det er en helt separat enhed, der kun bruges i nødsituationer og er ikke en del af lægemiddelgodkendelsesprocessen. (s. 28)
At opsummere:
Det er usandsynligt, at processen med at tildele et produkt EUA genererer beviser for sikkerhed eller effektivitet. Når først et produkt er tildelt EUA og administreret til patienter, er det usandsynligt, at der vil blive indhentet brugbar information til gavn for fremtidige patienter, fordi der ikke er nogen systematisk dataindsamling om effektivitet eller sikkerhed.
Baseret på alle disse meget klare oplysninger fra CDC/FDA og IMNA, ville det være rimeligt at konkludere, at nødbrugstilladelse er en proces, der bør anvendes meget velovervejet og kun i tilfælde af alvorlige nødsituationer.
Lad os nu se på, hvilke typer nødsituationer EUA er lovligt designet til at håndtere.
EAU er beregnet til WMD-nødsituationer
Lovene, der tillader EUA "Access Mechanism" beskrevet ovenfor, blev udarbejdet til tilfælde af ekstreme, øjeblikkelige nødsituationer, der involverer masseødelæggelsesvåben (WMD), også kaldet CBRN (kemiske, biologiske, radiologiske, nukleare) agenser.
Her er hvordan Food & Drug Administration (FDA) beskriver sine EUA-beføjelser:
Section 564 of the FD&C Act (21 USC 360bbb – 3) giver FDA mulighed for at styrke beskyttelsen af folkesundheden mod biologiske, kemiske, nukleare og radiologiske agenser.
Med denne EUA-autoritet kan FDA hjælpe med at sikre, at medicinske modforanstaltninger kan bruges i nødstilfælde til at diagnosticere, behandle eller forebygge alvorlige eller livstruende sygdomme eller tilstande forårsaget af biologiske, kemiske, nukleare eller radiologiske agenser, når der ikke er tilstrækkelige, godkendte , og tilgængelige alternativer (blandt andre kriterier).
Disse EUA-beføjelser blev givet i 2004 under meget specifikke omstændigheder i forbindelse med beredskab til angreb fra CBRN-agenter.
Som forklaret i Harvard Law's Bill of Health,
I sidste ende var det War on Terror, der ville give anledning til nødbrugstilladelse. Efter begivenhederne den 11. september 2001 og efterfølgende miltbrandpostangreb vedtog kongressen Project Bioshield Act af 2004.
optage indikerer, at kongressen var fokuseret på truslen om bioterror specifikt, ikke på at forberede sig på en naturligt forekommende pandemi.
I betragtning af en så snæver type virkelig ekstrem nødsituation, der involverer et WMD-angreb, er det forståeligt, hvorfor EUA "adgangsmekanismen" ikke kræver en masse regulatorisk tilsyn eller overholdelse af nogen standarder for fremstilling eller kliniske forsøg.
Så hvad kræver EUA-adgangsmekanismen egentlig?
De 3 trin til nødbrugsautorisation (EUA)
Der skal ske tre ting, for at EUA kan tildeles et medicinsk produkt:
- Secretary of Homeland Security, Secretary of Defense eller Secretary of Health and Human Services skal fastslå, at der er en nødsituation, der involverer et angreb eller en trussel om et angreb med et CBRN-agent eller en sygdom forårsaget af en sådan agent.
- FDA skal sikre sig, at den opfylder fire "lovpligtige kriterier", når den udsteder EUA.
- FDA skal "pålægge visse påkrævede betingelser" i EUA.
EUA Trin 1: Erklæring af en CBRN-nødsituation
Nøderklæringen for EUA er separat og ikke relateret til andre nøderklæringer, der kan udstedes af præsidenten, HHS-sekretæren eller nogen anden. Den skal udstedes specifikt med det formål at aktivere EUA og kan afsluttes eller forlænges uafhængigt af enhver anden nøderklæring.
Her er hvad siger EUA-loven er de fire mulige scenarier for aktivering af EUA "adgangsmekanismen:"
- en afgørelse fra Secretary of Homeland Security om, at der er en indenlandsk nødsituation eller et betydeligt potentiale for en indenlandsk nødsituation, der involverer en øget risiko for angreb med et eller flere biologiske, kemiske, radiologiske eller nukleare agenser;
- en afgørelse fra forsvarsministeren om, at der er en militær nødsituation eller et betydeligt potentiale for en militær nødsituation, der involverer en øget risiko for Forenet Stater militære styrker, herunder personel, der opererer under myndigheden i afsnit 10 eller afsnit 50, angreb med—
- et eller flere biologiske, kemiske, radiologiske eller nukleare agenser; eller
- en agent eller agenter, der kan forårsage eller på anden måde er forbundet med en overhængende livstruende og specifik risiko for United Stater militære styrker;
- en bestemmelse af Sekretær [of Health and Human Services], at der er en folkesundhedsnødsituation eller et betydeligt potentiale for en folkesundhedsnødsituation, der påvirker eller har et betydeligt potentiale til at påvirke den nationale sikkerhed eller sundheden og sikkerheden for Forenet Stater borgere, der bor i udlandet, og som involverer et eller flere biologiske, kemiske, radiologiske eller nukleare agenser eller en sygdom eller tilstand, der kan tilskrives sådanne agenser; eller
- identifikation af en væsentlig trussel i henhold til § 319F-2 i Lov om offentlig sundhedstjeneste [42 USC 247d–6b] tilstrækkelig til at påvirke den nationale sikkerhed eller sundhed og sikkerhed for Forenet Stater borgere, der bor i udlandet.
EUA Trin 2. Opfyldelse af de lovbestemte kriterier
Når en af sekretærerne har erklæret, at der er en nødsituation, der berettiger EUA, er der yderligere fire "lovpligtige kriterier", der skal opfyldes, for at FDA kan udstede EUA. Her er, hvordan FDA forklarer disse krav:
- Alvorlig eller livstruende sygdom eller tilstand
For at FDA kan udstede en EUA, skal CBRN-agenserne, der henvises til i HHS-sekretærens EUA-erklæring, være i stand til at forårsage en alvorlig eller livstruende sygdom eller tilstand.
- Bevis på effektivitet
Medicinske produkter, der kan komme i betragtning til en EUA, er dem, der "kan være effektive" til at forebygge, diagnosticere eller behandle alvorlige eller livstruende sygdomme eller tilstande, der kan være forårsaget af en eller flere CBRN-agenser identificeret i HHS-sekretærens erklæring om nødsituation eller trussel om nødsituation i henhold til paragraf 564(b).
Standarden "kan være effektiv" for EUA'er giver et lavere bevisniveau end den "effektivitet"-standard, som FDA bruger til produktgodkendelser. FDA har til hensigt at vurdere den potentielle effektivitet af et muligt EUA-produkt fra sag til sag ved hjælp af en risiko-benefit-analyse, som forklaret nedenfor.
[BOLDANSIGT TILFØJET]
- Risiko-Benefit Analyse
Et produkt kan komme i betragtning til en EUA, hvis kommissæren fastslår, at de kendte og potentielle fordele ved produktet, når det bruges til at diagnosticere, forebygge eller behandle den identificerede sygdom eller tilstand, opvejer de kendte og potentielle risici ved produktet.
Ved at afgøre, om de kendte og potentielle fordele ved produktet opvejer de kendte og potentielle risici, har FDA har tænkt sig at kigge på det samlede videnskabelige bevis for at foretage en overordnet risiko-benefit-bestemmelse. Sådanne beviser, som kunne opstå fra forskellige kilder, kan omfatte (men er ikke begrænset til): resultater af indenlandske og udenlandske kliniske forsøg, in vivo-effektivitetsdata fra dyremodeller og in vitro-data, tilgængelig for FDA-betragtning. FDA vil også vurdere kvaliteten og kvantiteten af tilgængelige beviseri betragtning af den nuværende videnskabelige viden.
[BOLDANSIGT TILFØJET]
- Ingen alternativer
For at FDA kan udstede en EUA, må der ikke være noget passende, godkendt og tilgængeligt alternativ til kandidatproduktet til diagnosticering, forebyggelse eller behandling af sygdommen eller tilstanden. Et potentielt alternativt produkt kan betragtes som "utilgængeligt", hvis der ikke er tilstrækkelige forsyninger af det godkendte alternativ til fuldt ud at opfylde nødbehovet.
EUA Trin 3. Indførelse af de påkrævede betingelser
Når først vi har den EUA-specifikke nøddeklaration, og når FDA har fastslået, at produktet kan være effektivt, og at uanset tilgængelig dokumentation viser, at fordelene opvejer dets risici, er der endnu et lag af relateret regulering.
Her er hvordan en 2018 Congressional Research Service rapport om EUA forklarer dette:
FFDCA §564 pålægger FDA at pålægge visse påkrævede betingelser i en EUA og giver mulighed for yderligere skønsmæssige betingelser, hvor det er relevant. De påkrævede betingelser varierer afhængigt af, om EUA er for et ikke-godkendt produkt eller for en ikke-godkendt brug af et godkendt produkt. For et ikke-godkendt produkt skal brugsbetingelserne:
(1) sikre, at sundhedspersonale, der administrerer produktet, modtager den nødvendige information;
(2) sikre, at personer, som produktet administreres til, modtager den nødvendige information;
(3) sørge for overvågning og rapportering af uønskede hændelser forbundet med produktet; og
(4) sørge for registrering og rapportering fra fabrikantens side.
Konklusion
Som bemærket i denne artikel anerkender FDA/CDC klart, at processen med at give Emergency Use Authorization (EUA) sandsynligvis ikke vil generere nogen information om effektiviteten eller sikkerheden af et produkt. Når vi ser på bogstavet i loven om EUA, ser vi, at dette faktisk er en korrekt vurdering.
EUA-loven pålægger ingen juridiske eller regulatoriske standarder, der kan afgøre, om et produkt er sikkert eller effektivt. De eneste standarder er, om FDA mener, at produktet kan være effektivt, og at dets kendte fordele opvejer dets kendte skader. Hvis der ikke er nogen kendte skader eller kendte fordele, fordi produktet aldrig har været igennem lægemiddelgodkendelsesprocessen, kan FDA bruge den information eller de standarder, den vælger til at træffe denne bestemmelse.
Det følger af alt dette, at en virksomhed, hvis produkt er en kandidat til EUA, kan forsøge at demonstrere produktets sikkerhed og/eller effektivitet på alle måder, den vælger. Eksistensen af et sådant forsøg (uanset om det er et klinisk forsøg eller en anden dataindsamlingsmekanisme), og hvordan dette forsøg udføres, er alt op til virksomheden. Intet i EUA-loven gælder for, hvordan virksomheden designer, udfører eller analyserer undersøgelser eller andre dataindsamlingsmekanismer, den vælger at forfølge.
Anvendt på Covid-produkter betyder dette:
- Ingen sikkerheds- eller effektdata fra kliniske forsøg var påkrævet, for at Covid-produkter kunne modtage EUA.
- Eventuelle kliniske forsøg, der henvises til i EUA-processen, blev udført uden lovligt gældende regulatoriske standarder.
- Når vi finder ud af, at disse produkter mangler effektivitet eller sikkerhed, er det ikke en overraskelse. Det er et meget sandsynligt resultat af processen.
- Der er ingen data fra EUA-processen, som kan baseres på ikke-EUA-beslutninger om produktets sikkerhed eller effektivitet. Så enhver ikke-EUA brug af produktet vil kræve at gennemgå den juridiske godkendelsesproces for almindelige medicinske produkter fra begyndelsen.
Mere om godkendelsesprocessen for Covid-vacciner link..
Genudgivet fra forfatterens understak
Udgivet under a Creative Commons Attribution 4.0 International licens
For genoptryk, sæt venligst det kanoniske link tilbage til originalen Brownstone Institute Artikel og forfatter.








